В 1885 году французским физиком и химиком Ле Шателье был выведен, а в 1887 году немецким физиком Брауном обоснован закон химического равновесия и константа химического равновесия, а также изучена их зависимость от влияния различных внешних факторов.
Суть химического равновесия
Равновесие - состояние, означающее, что вещи всегда движутся. Продукты разлагаются на реактивы, а реактивы объединяются в продукты. Вещи движутся, но концентрации остаются неизменными. Реакция записывается с двойной стрелкой вместо знака равенства, чтобы показать, что она обратима.

Классические закономерности
Еще в прошлом веке химиками были открыты определенные закономерности, которые предусматривают вероятность изменения направления реакции в одной и той же емкости. Знания о том, как протекают химические реакции, невероятно важны, как для лабораторных исследований, так и промышленного производства. При этом большое значение имеет возможность контролировать все эти явления. Человеку свойственно вмешиваться во многие природные процессы, в особенности это касается обратимых, чтобы потом использовать их себе на благо. От знаний о химических реакциях будет больше пользы, если в совершенстве владеть рычагами управления ими.
Закон действующих масс в химии использую химики, чтобы правильно рассчитывать скорости протекания реакций. Он дает четкое представление о том, что ни один не будет доведен до конца в случае, если он будет проходить в системе закрытого типа. Молекулы образующихся веществ находятся в постоянном и беспорядочном движении, и может вскоре возникнуть обратная реакция, при которой будут восстанавливаться молекулы исходного материала.
В промышленности чаще всего используют открытые системы. Сосуды, аппараты и другие емкости, где проходят химические реакции, остаются незапертыми. Это необходимо для того, чтобы во время этих процессов можно было извлечь нужный продукт и избавиться от бесполезных продуктов реакции. Например, уголь сжигается в открытых топках, цемент производится в печах открытого типа, домны функционируют при постоянной подаче воздуха, а аммиак синтезируется при непрерывном удалении самого аммиака.

Обратимые и необратимые химические реакции
Исходя из названия, можно дать соответствующие определения: необратимыми считаются реакции, доводимые до конца, не изменяющие своего направления и протекающие по заданной траектории вне зависимости от перепадов давления и температурных колебаний. Их отличительной особенностью является то, что некоторые продукты могут покидать сферу реакции. Таким образом, например, можно получить газ (CaCO 3 = CaO + CO 2), осадок (Cu(NO 3) 2 + H 2 S = CuS + 2HNO 3) или другие также будет считаться необратимой, если во время процесса выделяется большое количество тепловой энергии, например: 4P + 5O 2 = 2P 2 O 5 + Q.
Практически все реакции, которые происходят в природе, являются обратимыми. Независимо от таких внешних условий, как давление и температура, практически все процессы могут протекать одновременно в разных направлениях. Как гласит закон действующих масс в химии, количество поглощенной теплоты будет равно количеству выделенной, это значит, что если одна реакция была экзотермической, то вторая (обратная) буде эндотермической.
Химическое равновесие: константа химического равновесия
Реакции - это «глаголы» химии - деятельность, которую изучают химики. Многие реакции переходят к их завершению, а затем останавливаются, а это означает, что реагенты полностью преобразуются в продукты, не имея возможности вернуться в исходное состояние. В некоторых случаях реакция действительно необратима, например, когда сжигание изменяет как физические, так и химические Однако существует множество других обстоятельств, в которых является не только возможной, но и непрерывной, так как продукты первой реакции становятся реагентами во второй.
Динамическое состояние, в котором концентрации реагентов и продуктов остаются постоянными, называется равновесием. Можно предсказать поведение веществ с помощью определенных законов, которые применяются в отраслях, стремящихся снизить издержки производства конкретных химических веществ. Для понимания процессов, которые сохраняют или потенциально угрожают здоровью людей, также полезным является понятие химического равновесия. Константа химического равновесия представляет собой значение фактора реакции, которое зависит от ионной силы и температуры, и не зависит от концентраций реагентов и продуктов в растворе.

Вычисление константы равновесия
Эта величина является безразмерной, то есть не имеющей определенного количества единиц. Хотя расчет обычно записывается для двух реагентов и двух продуктов, он работает для любого количества участников реакции. Расчет и интерпретация константы равновесия зависят от того, связана ли химическая реакция с однородным или гетерогенным равновесием. Это значит, что все вступающие в реакцию компоненты могут быть чистыми жидкостями или газами. Для реакций, которые достигают гетерогенного равновесия, присутствует, как правило, не одна фаза, а как минимум две. Например, жидкости и газы или и жидкости.

Значение константы равновесия
Для любой заданной температуры для константы равновесия существует только одно значение, которое изменяется только в том случае, если температура, при которой происходит реакция, изменяется в ту или иную сторону. Можно сделать некоторые прогнозы относительно химической реакции, исходя из того, является ли постоянная равновесия большой или малой. Если значение очень велико, то равновесие благоприятствует реакции вправо и получается больше продуктов, чем было реагентов. Реакцию в этом случае можно назвать «полной» или «количественной».
Если значение константы равновесия невелико, то оно благоприятствует реакции влево, где количество реагентов было больше, чем образовавшихся продуктов. Если это значение стремится к нулю, можно считать, что реакция не возникает. Если же значения константы равновесия для прямой и обратной реакции почти одинаковы, то количество реагентов и продуктов будет тоже почти одинаковым. Этот тип реакции считается обратимым.

Рассмотрим конкретную обратимую реакцию
Возьмем таких два химических элемента, как йод и водород, которые при смешивании дают новое вещество - иодоводород.
За v 1 примем скорость прямой реакции, за v 2 - скорость обратной реакции, k - константа равновесия. Используя закон действия масс, получаем следующее выражение:
v 1 = k 1 * c(H 2) * c(I 2),
v 2 = k 2 * c 2 (HI).
При смешивании молекул йода (I 2) и водорода (H 2) начинается их взаимодействие. На начальном этапе концентрация этих элементов максимальная, а вот к концу реакции максимальной будет концентрация нового соединения - иодоводорода (HI). Соответственно, разными будут и скорости реакций. В самом начале они будут максимальными. Со временем наступает момент, когда эти значения будут равными, он и является состоянием, которое называется химическим равновесием.
Выражение константы химического равновесия, как правило, обозначают с применением квадратных скобок: , , . Так как при состоянии равновесия скорости равны, то:
k 1 = k 2 2 ,
так получаем уравнение константы химического равновесия:
k 1 /k 2 = 2 / = K.

Принцип Ле Шателье-Брауна
Существует следующая закономерность: если на систему, которая находится в равновесии, произвести определенное воздействие (изменить условия химического равновесия путем изменения температуры или давления, например), то баланс будет смещаться, чтобы частично противодействовать эффекту изменения. В дополнение к химии этот принцип также применим в несколько разных формах к областям фармакологии и экономики.

Константа химического равновесия и способы ее выражения
Равновесное выражение может быть выражено в терминах концентрации продуктов и реагентов. Только химические вещества в водной и газообразной фазах включены в равновесную формулу, поскольку концентрации жидкостей и твердых веществ не изменяются. Какие факторы влияют на химическое равновесие? Если в нем участвует чистая жидкость или твердое вещество, считается, что оно имеет К= 1, и соответственно перестает браться в расчет, за исключением высококонцентрированных растворов. Например, чистая вода имеет активность 1.
Другим примером является твердый углерод, который может образовываться реакцией двух молекул монооксида карбона с образованием углекислого газа и углерода. Факторы, которые могут повлиять на равновесие, включают в себя добавление реагента или продукта (изменение концентрации влияет на баланс). Добавление реагента может привести к равновесию справа в химическом уравнении, где появляется больше форм продукта. Добавление продукта может привести к равновесию слева, так как больше становится форм реагентов.

Равновесие возникает, когда реакция, проходящая в обоих направлениях, имеет неизменное соотношение продуктов и реагентов. В целом, химическое равновесие статично, так как количественное соотношение продуктов и реагентов постоянны. Однако более пристальный взгляд показывает, что равновесие на самом деле является очень динамичным процессом, так как реакция движется в обоих направлениях в равном темпе.
Динамическое равновесие является примером функции устойчивого состояния. Для системы в устойчивом состоянии наблюдаемое в настоящее время поведение продолжается и в будущем. Поэтому, как только реакция достигнет равновесия, отношение концентраций продукта и реагента останется таким же, хотя реакция продолжается.

Как просто рассказать о сложном?
Такие понятия, как химическое равновесие и константа химического равновесия, являются достаточно сложными для понимания. Возьмем пример из жизни. Вы когда-нибудь застревали на мосту между двумя городами и обращали внимание на то, что движение в другом направлении плавное и размеренное, в то время как вы безнадежно застряли в пробке? Это нехорошо.
Что, если бы машины размеренно и с одинаковой скоростью двигались с обеих сторон? Оставалось бы количество автомобилей в обоих городах постоянным? Когда скорость въезда и выезда в оба города одинакова, а количество автомобилей в каждом городе стабильно с течением времени, это означает, что весь процесс находится в динамическом равновесии.
Химическим равновесием называется такое состояние обратимой химической реакции
aA + b B = c C + d D,
при котором с течением времени не происходит изменения концентраций реагирующих веществ в реакционной смеси. Состояние химического равновесия характеризуется константой химического равновесия :
где C i – концентрации компонентов в равновесной идеальной смеси.
Константа равновесия может быть выражена также через равновесные мольные доли X i компонентов:
Для реакций, протекающих в газовой фазе, константу равновесия удобно выражать через равновесные парциальные давления P i компонентов:
Для идеальных газов P i = C i RT и P i = X i P , где P – общее давление, поэтому K P , K C и K X связаны следующим соотношением:
K P = K C (RT) c+d–a–b = K X P c+d–a–b . (9.4)
Константа равновесия связана с r G o химической реакции:
![]() (9.5)
(9.5)
![]() (9.6)
(9.6)
Изменение r G или r F в химической реакции при заданных (не обязательно равновесных) парциальных давлениях P i или концентрациях C i компонентов можно рассчитать по уравнению изотермы химической реакции (изотермы Вант-Гоффа ):
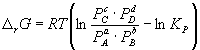 . (9.7)
. (9.7)
 . (9.8)
. (9.8)
Согласно принципу Ле Шателье , если на систему, находящуюся в равновесии, оказать внешнее воздействие, то равновесие сместится так, чтобы уменьшить эффект внешнего воздействия. Так, повышение давления сдвигает равновесие в сторону уменьшения количества молекул газа. Добавление в равновесную смесь какого-либо компонента реакции сдвигает равновесие в сторону уменьшения количества этого компонента. Повышение (или понижение) температуры сдвигает равновесие в сторону реакции, протекающей с поглощением (выделением) теплоты.
Количественно зависимость константы равновесия от температуры описывается уравнением изобары химической реакции (изобары Вант-Гоффа )
![]() (9.9)
(9.9)
и изохоры химической реакции (изохоры Вант-Гоффа )
![]() . (9.10)
. (9.10)
Интегрирование уравнения (9.9) в предположении, что r H реакции не зависит от температуры (что справедливо в узких интервалах температур), дает:
![]() (9.11)
(9.11)
![]() (9.12)
(9.12)
где C – константа интегрирования. Таким образом, зависимость ln K P от 1/Т должна быть линейной, а наклон прямой равен – r H /R .
Интегрирование в пределах K 1 , K 2 , и T 1, T 2 дает:
 (9.13)
(9.13)
 (9.14)
(9.14)
По этому уравнению, зная константы равновесия при двух разных температурах, можно рассчитать r H реакции. Соответственно, зная r H реакции и константу равновесия при одной температуре, можно рассчитать константу равновесия при другой температуре.
ПРИМЕРЫ
CO(г) + 2H 2 (г) = CH 3 OH(г)
при 500 K. f G o для CO(г) и CH 3 OH(г) при 500 К равны –155.41 кДж. моль –1 и –134.20 кДж. моль –1 соответственно.
Решение. G o реакции:
r G o = f G o (CH 3 OH) – f G o (CO) = –134.20 – (–155.41) = 21.21 кДж. моль –1 .
![]() = 6.09 10 –3 .
= 6.09 10 –3 .
Пример 9-2. Константа равновесия реакции
равна K P = 1.64 10 –4 при 400 o C. Какое общее давление необходимо приложить к эквимолярной смеси N 2 и H 2 , чтобы 10% N 2 превратилось в NH 3 ? Газы считать идеальными.
Решение. Пусть прореагировало моль N 2 . Тогда
| N 2 (г) | + | 3H 2 (г) | = | 2NH 3 (г) | |
| Исходное количество | 1 | 1 | |||
| Равновесное количество | 1– | 1–3 | 2 (Всего: 2–2) | ||
| Равновесная мольная доля: |
Следовательно, K
X = 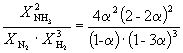 и K P = K X . P
–2
=
и K P = K X . P
–2
= 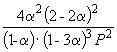 .
.
Подставляя = 0.1 в полученную формулу, имеем
1.64 10 –4 =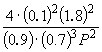 , откуда P
= 51.2 атм.
, откуда P
= 51.2 атм.
Пример 9-3. Константа равновесия реакции
CO(г) + 2H 2 (г) = CH 3 OH(г)
при 500 K равна K P = 6.09 10 –3 . Реакционная смесь, состоящая из 1 моль CO, 2 моль H 2 и 1 моль инертного газа (N 2) нагрета до 500 K и общего давления 100 атм. Рассчитать состав равновесной смеси.
Решение. Пусть прореагировало моль CO. Тогда
| CO(г) | + | 2H 2 (г) | = | CH 3 OH(г) | |
| Исходное количество: | 1 | 2 | 0 | ||
| Равновесное количество: | 1– | 2–2 | |||
| Всего в равновесной смеси: | 3–2 моль компонентов + 1 моль N 2 = 4–2 моль | ||||
| Равновесная мольная доля | |||||
Следовательно, K
X = 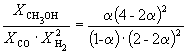 и K P = K X . P –2
=
и K P = K X . P –2
= ![]() .
.
Таким образом, 6.09 10 –3
= ![]() .
.
Решая это уравнение, получаем = 0.732. Соответственно, мольные доли веществ в равновесной смеси равны: = 0.288, = 0.106, = 0.212 и = 0.394.
Пример 9-4. Для реакции
N 2 (г) + 3H 2 (г) = 2NH 3 (г)
при 298 К K P = 6.0 10 5 , а f H o (NH 3) = –46.1 кДж. моль –1 . Оценить значение константы равновесия при 500 К.
Решение. Стандартная мольная энтальпия реакции равна
r H o = 2 f H o (NH 3) = –92.2 кДж. моль –1 .
Согласно уравнению (9.14),  =
=
Ln (6.0 10 5) + ![]() = –1.73, откуда K
2 =
0.18.
= –1.73, откуда K
2 =
0.18.
Отметим, что константа равновесия экзотермической реакции уменьшается с ростом температуры, что соответствует принципу Ле Шателье.
ЗАДАЧИ
- При 1273 К и общем давлении 30 атм в равновесной смеси
- При 2000 o C и общем давлении 1 атм 2% воды диссоциировано на водород и кислород. Рассчитать константу равновесия реакции
- Константа равновесия реакции
- Константа равновесия реакции
- Сосуд объемом 3 л, содержащий 1.79 10 –2 моль I 2 , нагрели до 973 K. Давление в сосуде при равновесии оказалось равно 0.49 атм. Считая газы идеальными, рассчитать константу равновесия при 973 K для реакции
- Для реакции
- Для реакции
- Сосуд объемом 1 л, содержащий 0.341 моль PCl 5 и 0.233 моль N 2 , нагрели до 250 o C. Общее давление в сосуде при равновесии оказалось равно 29.33 атм. Считая все газы идеальными, рассчитать константу равновесия при 250 o C для протекающей в сосуде реакции
- Константа равновесия реакции
- При 25 o C f G o (NH 3) = –16.5 кДж. моль –1 . Рассчитать r G реакции образования NH 3 при парциальных давлениях N 2 , H 2 и NH 3 , равных 3 атм, 1 атм и 4 атм соответственно. В какую сторону реакция будет идти самопроизвольно при этих условиях?
- Экзотермическая реакция
- Константа равновесия газофазной реакции изомеризации борнеола (C 10 H 17 OH) в изоборнеол равна 0.106 при 503 K. Смесь 7.5 г борнеола и 14.0 г изоборнеола поместили в сосуд объемом 5 л и выдерживали при 503 K до достижения равновесия. Рассчитать мольные доли и массы борнеола и изоборнеола в равновесной смеси.
- Равновесие в реакции
- Рассчитать общее давление, которое необходимо приложить к смеси 3 частей H 2 и 1 части N 2 , чтобы получить равновесную смесь, содержащую 10% NH 3 по объему при 400 o C. Константа равновесия для реакции
- При 250 o C и общем давлении 1 атм PCl 5 диссоциирован на 80% по реакции
- При 2000 o C для реакции
- Рассчитать стандартную энтальпию реакции, для
которой константа равновесия
а) увеличивается в 2 раза, б) уменьшается в 2 раза при изменении температуры от 298 К до 308 К. - Зависимость константы равновесия реакции 2C 3 H 6 (г) = C 2 H 4 (г) + C 4 H 8 (г) от температуры между 300 К и 600 К описывается уравнением
CO 2 (г) + C(тв) = 2CO(г)
содержится 17% (по объему) CO 2 . Сколько процентов CO 2 будет содержаться в газе при общем давлении 20 атм? При каком давлении в газе будет содержаться 25% CO 2 ?
H 2 O(г) = H 2 (г) + 1/2O 2 (г) при этих условиях.
CO(г) + H 2 O(г) = CO 2 (г) + H 2 (г)
при 500 o C равна K p = 5.5. Смесь, состоящая из 1 моль CO и 5 моль H 2 O, нагрели до этой температуры. Рассчитать мольную долю H 2 O в равновесной смеси.
N 2 O 4 (г) = 2NO 2 (г)
при 25 o C равна K p = 0.143. Рассчитать давление, которое установится в сосуде объемом 1 л, в который поместили 1 г N 2 O 4 при этой температуре.
I 2 (г) = 2I (г).
при 250 o C r G o = –2508 Дж. моль –1 . При каком общем давлении степень превращения PCl 5 в PCl 3 и Cl 2 при 250 o C составит 30%?
2HI(г) = H 2 (г) + I 2 (г)
константа равновесия K P = 1.83 10 –2 при 698.6 К. Сколько граммов HI образуется при нагревании до этой температуры 10 г I 2 и 0.2 г H 2 в трехлитровом сосуде? Чему равны парциальные давления H 2 , I 2 и HI?
PCl 5 (г) = PCl 3 (г) + Cl 2 (г)
CO(г) + 2H 2 (г) = CH 3 OH(г)
при 500 K равна K P = 6.09 10 –3 . Рассчитать общее давление, необходимое для получения метанола с 90% выходом, если CO и H 2 взяты в соотношении 1: 2.
CO(г) + 2H 2 (г) = CH 3 OH(г)
находится в равновесии при 500 K и 10 бар. Если газы идеальные, как повлияют на выход метанола следующие факторы: а) повышение T ; б) повышение P ; в) добавление инертного газа при V = const; г) добавление инертного газа при P = const; д) добавление H 2 при P = const?
2NOCl(г) = 2NO(г) + Cl 2 (г)
устанавливается при 227 o C и общем давлении 1.0 бар, когда парциальное давление NOCl равно 0.64 бар (изначально присутствовал только NOCl). Рассчитать r G o для реакции. При каком общем давлении парциальное давление Cl 2 будет равно 0.10 бар?
N 2 (г) + 3H 2 (г) = 2NH 3 (г)
при 400 o C равна K = 1.60 10 –4 .
PCl 5 (г) = PCl 3 (г) + Cl 2 (г).
Чему будет равна степень диссоциации PCl 5 , если в систему добавить N 2 , чтобы парциальное давление азота было равно 0.9 атм? Общее давление поддерживается равным 1 атм.
N 2 (г) + O 2 (г) = 2NO(г)
K p = 2.5 10 –3 . В равновесной смеси N 2 , O 2 , NO и инертного газа при общем давлении 1 бар содержится 80% (по объему) N 2 и 16% O 2 . Сколько процентов по объему составляет NO? Чему равно парциальное давление инертного газа?
ln K = –1.04 –1088 /T +1.51 10 5 /T 2 .
Константа равновесия
Конста́нта равнове́сия - величина, определяющая для данной химической реакции соотношение между термодинамическими активностями (либо, в зависимости от условий протекания реакции, парциальными давлениями , концентрациями или фугитивностями) исходных веществ и продуктов в состоянии химического равновесия (в соответствии с законом действующих масс). Зная константу равновесия реакции, можно рассчитать равновесный состав реагирующей смеси, предельный выход продуктов, определить направление протекания реакции.
Способы выражения константы равновесия
Например, для реакции окисления монооксида углерода :
2CO + O 2 = 2CO 2
константа равновесия может быть рассчитана по уравнению:
где Δn - изменение числа молей веществ в ходе реакции. Видно, что K x зависит от давления. Если число молей продуктов реакции равно числу молей исходных веществ (), то .
Стандартная константа равновесия
Стандартная константа равновесия реакции в смеси идеальных газов (когда начальные парциальные давления участников реакции равны их значениям в стандартном состоянии = 0,1013 МПа или 1 атм) может быть рассчитана по выражению:
где - относительные парциальные давления компонентов, .Стандартная константа равновесия - безразмерная величина. Она связана с K p соотношением:
Видно, что если выражены в атмосферах, то и .
Для реакции в смеси реальных газов в стандартном начальном состоянии парциальные фугитивности газов принимаются равными их парциальным давлениям = 0,1013 МПа или 1 атм. K f связана с K 0 соотношением:
где γ i - коэффициент фугитивности i-го реального газа в смеси.Константа равновесия реакций в гетерогенных системах
FeO т + CO г = Fe т + CO 2гконстанта равновесия (при условии, что газовая фаза идеальна) имеет вид:
Константа равновесия и изменение энергии Гиббса
Константа равновесия и константа скорости реакции
Для обратимой химической реакции константа равновесия может быть выражена через константы скорости прямых и обратных реакций, исходя из того факта, что в состоянии равновесия скорости прямой и обратной реакций равны. Например, для элементарной обратимой химической реакции первого порядка
где k 1 - константа скорости прямой реакции, а k 2 - обратной. Это важное соотношение даёт одну из «точек соприкосновения» химической кинетики и химической термодинамики.Методы расчета константы равновесия
Расчётные методы определения константы равновесия реакции обычно сводятся к вычислению тем или иным способом стандартного изменения энергии Гиббса в ходе реакции (ΔG 0 ), а затем использованию формулы:
, где - универсальная газовая постоянная .При этом следует помнить, что энергия Гиббса - функция состояния системы, то есть она не зависит от пути процесса, от механизма реакции, а определяется лишь начальным и конечным состояниями системы. Следовательно, если непосредственное определение или расчёт ΔG 0 для некоторой реакции по каким-либо причинам затруднены, можно подобрать такие промежуточные реакции, для которых ΔG 0 известно или может быть легко определено, и суммирование которых даст рассматриваемую реакцию (см. Закон Гесса). В частности, в качестве таких промежуточных реакций часто используют реакции образования соединений из элементов.
Энтропийный расчёт изменения энергии Гиббса и константы равновесия реакции
Энтропийный метод расчёта ΔG реакции является одним из самых распространённых и удобных . Он основан на соотношении:
или, соответственно, для стандартного изменения энергии Гиббса:
Здесь ΔH 0 при постоянных давлении и температуре равно тепловому эффекту реакции, методы расчёта и экспериментального определения которого известны - см., например, уравнение Кирхгофа :
Необходимо получить изменение энтропии в ходе реакции. Эта задача может быть решена несколькими способами, например:
- По термическим данным - с опорой на тепловую теорему Нернста и с использованием сведений о температурной зависимости теплоёмкости участников реакции. Например, для веществ, при нормальных условиях находящихся в твёрдом состоянии:
Итак, если извеcтны , и температурные зависимости теплоёмкости, может быть рассчитано по формуле:
Несколько упрощённый вариант этой формулы получают, считая сумму теплоёмкостей веществ не зависящей от температуры и равной сумме теплоёмкостей при 298 K:
И еще более упрощённый расчёт проводят, приравнивая сумму теплоёмкостей к нулю:
Переход от к константе равновесия осуществляется по приведённой выше формуле.
Расчёт константы равновесия методами статистической термодинамики
| Этот раздел статьи ещё не написан. |
п о с т о я н н а я (от лат. constans, род. п. constantis – постоянный, неизмененный), – такой из объектов в нек-рой теории, значение к-рого в рамках этой теории (или, иногда, более узкого рассмотрения) считается всегда одним и тем же. К. противопоставляются таким объектам, значения к-рых изменяются (сами по себе или в зависимости от изменения значений др. объектов). Наличие К. при выражении мн. законов природы и общества отражает относит. неизменность тех или иных сторон реальной действительности, проявляющуюся в наличии закономерностей. Важной разновидностью К. является К., относящиеся к числу физич. величин, – таких, как длина, время, сила, масса (напр., масса покоя электрона), или более сложных величин, численно выразимых через отношения между этими К. или их степенями, – таких, как объем, скорость, работа и т.п. (напр., ускорение силы тяжести у поверхности Земли). Те из К. этого рода, к-рые считаются в совр. физике (в рамках соответствующих ее теорий) имеющими значение для всей наблюдаемой части Вселенной, наз. мировыми (или универсальными) К.; примерами таких К. являются скорость света в пустоте, квантовая постоянная Планка (т.е. величина т.н. кванта действия), гравитационная постоянная и др. На большое значение мировых К. наука обратила внимание в 20–30-х гг. 20 в. При этом нек-рые зарубежные ученые (англ. физик и астроном А. Эддингтон, нем. физик Гейзенберг, австр. физик А. Марх и др.) пытались дать им идеалистич. истолкование. Так, Эддингтон видел в системе мировых К. одно из проявлений самостоят. существования идеальных математич. форм, выражающих гармонию природы и ее законов. На самом же деле универсальные К. отражают не мнимое самостоят. бытие (вне вещей и познания) указанных форм, а (выражаемые обычно математически) фундаментальные законо-мерности объективной действительности, в частности закономерности, связанные со строением материи. Глубокий диалектич. смысл мировых К. раскрывается в том, что нек-рые из них (квантовая постоянная Планка, скорость света в пустоте) являются своего рода масштабами, разграничивающими различные классы процессов, протекающих принципиально по-разному; вместе с тем такие К. указывают и на наличие определ. связи между явлениями этих классов. Так, связь между законами классич. и релятивистской механики (см. Относительности теория) может быть установлена из рассмотрения такого предельного перехода уравнений движения релятивистской механики в уравнения движения классич. механики, к-рый связан с идеализацией, состоящей в отказе от представления о скорости света в пустоте как о конечной К. и в понимании скорости света как бесконечно большой; при др. идеализации, состоящей в рассмотрении кванта действия как бесконечно малой величины, уравнения движения квантовой теории переходят в уравнения движения классич. механики и т.п. Кроме этих важнейших К., определяемых сугубо физически и фигурирующих в формулировках многих осн. законов природы, широко используются там же и такие, определяемые чисто математически, К., как числа 0; 1; ? (отношение длины окружности к диаметру); е (основание натуральных логарифмов); постоянная Эйлера и др. Не менее часто используются и К., к-рые являются результатами известных математич. операций над указанными К. Но чем труднее выразить часто употребляемую К. через более просто определяемые К. (или такие самые простые К., как 0 и 1) и известные операции, тем более самостоятельным является ее участие в формулировках тех законов и соотношений, в к-рых она встречается, тем чаще для нее вводят спец. обозначение, вычисляют или измеряют ее возможно точнее. Иные из величин встречаются эпизодически и являются К. лишь в рамках рассмотрения нек-рой задачи, причем они могут даже зависеть от выбора условий (значений параметров) задачи, становясь К. лишь при фиксировании этих условий. Такие К. часто обозначают буквами С или K (не связывая эти обозначения раз навсегда с одной и той же К.) или просто пишут, что такая-то величина = const. А. Кузнецов, И. Ляхов. Москва. В тех случаях, когда в математике или логике роль рассматриваемых объектов играют функции, К. называются такие из них, значение к-рых не зависит от значений аргументов этих функций. Напр., К. является разность х–х как функция от х, т.к. при всех (числовых) значениях переменной х значением функции х–х является одно и то же число 0. Примером функции алгебры логики, являющейся К., является A/A (рассматриваемая как функция от "переменного высказывания" А), т.к. она при всех возможных значениях своего аргумента А имеет (в рамках обычной, классич. алгебры логики) одно и то же значение 1 (к-рым характеризуется условно отождествляемое с ним логич. значение "истина"). Примером более сложной К. из алгебры логики является функция (АВ?ВА). В нек-рых случаях функция, значение к-рой постоянно, отождествляется с самим этим значением. При этом значение функции выступает уже как К. (точнее, как функция, являющаяся К.). Аргументами этой функции могут считаться любые выбранные буквенные переменные (напр., А, В, х, у и т.п.), т.к. все равно она от них не зависит. В др. случаях такого отождествления функции, являющейся К., с ее значением не производят, т.е. различают такие две К., у одной из к-рых среди ее аргументов есть переменная, к-рой нет у другой. Это позволяет, напр., определять функцию как ее таблицу, а также упрощает схематич. определение нек-рых операций над функциями. Наряду с такими К., значения к-рых являются числами (быть может и именованными) или характеризуются числами, встречаются и иные К. Напр., в множеств теории важной К. является натуральный ряд N, т.е. множество всех целых неотрицат. чисел. Значением функции, являющейся К., тоже может быть объект любой природы. Напр., рассматривая функции от такой переменной А, значениями к-рой являются подмножества натурального ряда, можно определить такую из этих функций, значением к-рой при всех значениях переменной А будет множество всех простых чисел. Кроме физич. величин и функций в роли таких объектов, нек-рые из к-рых оказываются К., часто (особенно в логике и семантике) рассматривают знаки и их комбинации: слова, предложения, термины, формулы и т.п., а в качестве значения тех из них, о значениях к-рых особо не говорится, их смысловые значения (если таковые имеются). При этом выявляются новые К. Так, в арифметич. выражении (терме) 2+3–2 К. оказываются не только числа 2 и 3 и результаты операций над ними, но также и знаки + и –, значениями к-рых являются операции сложения и вычитания. Эти знаки, являясь К. в рамках теоретич. рассмотрения обычных школьных арифметики и алгебры, перестают быть К., когда мы выходим в более широкую область совр. алгебры или логики, где знак + имеет в одних случаях значение операции обычного сложения чисел, в др. случаях (напр., в алгебре логики) – сложения по модулю 2 или булева сложения, в иных же случаях – иной операции. Однако при более узких рассмотрениях (напр., при построении конкретной алгебраич. или логич. системы) значения знаков операций фиксируются и эти знаки, в отличие от знаков переменных, становятся К. Выделение логич. К. играет особую роль в применении к объектам из естеств. языка. В роли логич. К. в рус. языке выступают, напр., такие союзы, как "и", "или" и др., такие кванторные слова, как "все", "всякий", "существует", "некоторый" и др., такие глаголы-связки, как "есть", "суть", "является" и др., а также такие более сложные словосочетания, как "если..., то", "если и только если", "существует единственный", "тот, который", "такой, что", "эквивалентно тому, что" и др. Средством выделения логич. К. в естеств. языке является усмотрение одинаковости их роли в огромном числе случаев умозаключений или иных рассуждений, позволяющее объединить эти случаи в ту или иную единую схему (логич. правило), в к-рой объекты, отличные от выделенных К., заменены соответствующими переменными. Чем меньшим числом схем удается охватить все рассматриваемые случаи рассуждений, чем проще сами эти схемы и чем больше мы гарантированы от возможности ошибочных рассуждений по ним, тем более оправданным является выбор фигурирующих в этих схемах логич. К. А. Кузнецов. Москва. Лит.: Эддингтон?., Пространство, время и тяготение, пер. с англ., О., 1923; Джинс Д., Вселенная вокруг нас, пер. с англ., Л.–М., 1932; Борн М., Таинственное число 137, в сб.: Успехи физ. наук, т. 16, вып. 6, 1936; Гейзенберг В., Филос. проблемы атомной физики, М., 1953; его же, Открытие Планка и осн. филос. вопросы учения об атомах, "Вопр. философии", 1958, No 11; его же, Физика и философия, М., 1963; Сб. ст. по матем. логике и ее приложения к нек-рым вопросам кибернетики, в кн.: Тр. матем. ин-та, т. 51, М., 1958; Кузнецов И. В., В чем прав и в чем ошибается Вернер Гейзенберг, "Вопр. философии", 1958, No 11; Успенский В. ?., Лекции о вычислимых функциях, М., 1960; Кэй Дж. и Лэби Т., Таблицы физ. и хим. постоянных, пер. с англ., 2 изд., М., 1962; Курош А. Г., Лекции по общей алгебре, М., 1962; Свидерский В. И., О диалектике элементов и структуры в объективном мире и в познании, М., 1962, гл. 3; ?ddington A. St., New pathways in science, Camb., 1935; его же, Relativity theory of protons and electrons, L., 1936; его же, The philosophy of physical science, N. Y.–Camb., 1939; Louis de Broglie, physicien et penseur, P., ; March ?., Die physikalische Erkenntnis und ihre Grenzen, 2 Aufl., Braunschweig, 1960.
K p = ∏ p i ν i {\displaystyle K_{p}=\prod p_{i}^{{\nu }_{i}}}
Например, для реакции окисления монооксида углерода :
2CO + O 2 = 2CO 2
константа равновесия может быть рассчитана по уравнению:
K p = p C O 2 2 p C O 2 ⋅ p O 2 {\displaystyle K_{p}={\frac {p_{CO_{2}}^{2}}{p_{CO}^{2}\cdot p_{O_{2}}}}} K p = K x P Δ n {\displaystyle K_{p}=K_{x}P^{\Delta n}}где Δn - изменение числа молей веществ в ходе реакции. Видно, что K x зависит от давления. Если число молей продуктов реакции равно числу молей исходных веществ ( Δ n = 0 {\displaystyle \Delta n=0} ), то K p = K x {\displaystyle K_{p}=K_{x}} .
Стандартная константа равновесия
Стандартная константа равновесия реакции в смеси идеальных газов (когда начальные парциальные давления участников реакции равны их значениям в стандартном состоянии = 0,1013 МПа или 1 атм) может быть рассчитана по выражению:
K 0 = ∏ (p i ~) v i {\displaystyle K^{0}=\prod ({\tilde {p_{i}}})^{v_{i}}} где p i ~ {\displaystyle {\tilde {p_{i}}}} - относительные парциальные давления компонентов, p i ~ = p i / p i 0 {\displaystyle {\tilde {p_{i}}}=p_{i}/p_{i}^{0}} .Стандартная константа равновесия - безразмерная величина. Она связана с K p соотношением:
K p = K 0 (p i 0) Δ n {\displaystyle K_{p}=K^{0}(p_{i}^{0})^{\Delta n}}Видно, что если p i 0 {\displaystyle p_{i}^{0}} выражены в атмосферах, то (p i 0) Δ n = 1 {\displaystyle (p_{i}^{0})^{\Delta n}=1} и K p = K 0 {\displaystyle K_{p}=K^{0}} .
Для реакции в смеси реальных газов в стандартном начальном состоянии парциальные фугитивности газов принимаются равными их парциальным давлениям f i 0 = p i 0 {\displaystyle f_{i}^{0}=p_{i}^{0}} = 0,1013 МПа или 1 атм. K f связана с K 0 соотношением:
K f = K 0 (γ i p i 0) Δ n {\displaystyle K_{f}=K^{0}(\gamma _{i}p_{i}^{0})^{\Delta n}} где γ i - коэффициент фугитивности i-го реального газа в смеси.Константа равновесия реакций в гетерогенных системах
FeO т + CO г = Fe т + CO 2гконстанта равновесия (при условии, что газовая фаза идеальна) имеет вид:
K p = p C O 2 p C O {\displaystyle K_{p}={\frac {p_{CO_{2}}}{p_{CO}}}}Термодинамическое описание равновесия
Наряду с обозначением Q для соотношения активностей веществ в произвольный момент реакции t ("коэффициент реакции ")
Q r = { S t } σ { T t } τ { A t } α { B t } β = ∏ a j (t) ν j ∏ a i (t) ν i = ∏ a n (t) ν n {\displaystyle Q_{r}={\frac {\left\{S_{t}\right\}^{\sigma }\left\{T_{t}\right\}^{\tau }}{\left\{A_{t}\right\}^{\alpha }\left\{B_{t}\right\}^{\beta }}}={\frac {\prod a_{j(t)}^{\nu _{j}}}{\prod a_{i(t)}^{\nu _{i}}}}=\prod a_{n(t)}^{\nu _{n}}} (обозначения для приведённой ниже реакции; последнее равенство написано в обозначении, что стехиометрические коэффициент берутся со знаком "+" для продуктов и со знаком "-" для исходных веществ)в химической термодинамике используется обозначение K eq для такого же по форме соотношения между равновесными активностями веществ
K e q = [ S ] σ [ T ] τ [ A ] α [ B ] β = ∏ a j (t = ∞) ν j ∏ a i (t = ∞) ν i = ∏ a n (t = ∞) ν n {\displaystyle K_{eq}={\frac {[S]^{\sigma }[T]^{\tau }}{[A]^{\alpha }[B]^{\beta }}}={\frac {\prod a_{j(t=\infty)}^{\nu _{j}}}{\prod a_{i(t=\infty)}^{\nu _{i}}}}=\prod a_{n(t=\infty)}^{\nu _{n}}} (то есть соотношения активностей в момент t = ∞ {\displaystyle t=\infty } , в момент равновесия). Далее приведено термодинамическое описание химического равновесия и описана связь K eq со стандартной энергией Гиббса процесса.В системе, где протекает химическая реакция
α A + β B ⇌ σ S + τ T {\displaystyle \alpha A+\beta B\rightleftharpoons \sigma S+\tau T}равновесие может быть описано условием
(d G d ξ) T , p = 0 {\displaystyle \left({\frac {dG}{d\xi }}\right)_{T,p}=0} где ξ {\displaystyle \xi } есть химическая переменнаяили, то же самое условие равновесия может быть записано с использованием химических потенциалов как
α μ A + β μ B = σ μ S + τ μ T {\displaystyle \alpha \mu _{A}+\beta \mu _{B}=\sigma \mu _{S}+\tau \mu _{T}}где химические потенциалы
μ A = μ A ⊖ + R T ln { A } {\displaystyle \mu _{A}=\mu _{A}^{\ominus }+RT\ln\{A\}} здесь {A} - строго говоря, активность реагента A; при допущениях об идеальных газах можно заменить их на давления, для реальных газов можно заменить на фугитивности, при допущении о том, что раствор подчиняется закону Генри , можно заменить на мольные доли , и при допущении, что раствор подчиняется закону Рауля - на парциальные давления ; для системы в равновесии может быть заменена на равновесную молярную концентрацию или на равновесную активность. Δ r G o = − R T ln K e q {\displaystyle \Delta _{r}G^{o}=-RT\ln K_{eq}}Равновесный состав смеси и направление реакции
Упомянутый выше "коэффициент реакции" Q (другие обозначения, встречающиеся в литературе - Ω {\displaystyle \Omega } или π {\displaystyle \pi } , "произведение реакции")
Q r = ∏ a n (t) ν n {\displaystyle Q_{r}=\prod a_{n(t)}^{\nu _{n}}}отражает соотношение текущих активностей всех участников реакции и может быть использован для определения направления реакции в момент, для которого известен Q
Если в момент t коэффициент Q > K, то текущие активности продуктов больше равновесных, и значит они должны уменьшиться к тому моменту, когда установится равновесие, то есть в данный момент протекает обратная реакция; Если Q = K, то равновесное состояние достигнуто и скорости прямой и обратной реакций равны; Если Q < K, то v 1 > v − 1 {\displaystyle v_{1}>v_{-1}}
С использованием величины Q r {\displaystyle Q_{r}} записывается уравнение изотермы химической реакции
Δ G p , T = R T ln Q r − R T ln K e q = R T ln Q r K e q = ∑ ν i μ i {\displaystyle \Delta G_{p,T}=RT\ln Q_{r}-RT\ln K_{eq}=RT\ln {\frac {Q_{r}}{K_{eq}}}=\sum \nu _{i}\mu _{i}}Где ν {\displaystyle \nu } - стехиометрические коэффициенты (для продуктов - со знаком "+", для исходных веществ - со знаком "-"; так же, как и в выражениях для Q и K), а μ {\displaystyle \mu } - химические потенциалы а стандартная энергия Гиббса и стандартная константа суть
Δ G p , T o = − R T ln K e q o = ∑ ν i μ i o {\displaystyle \Delta G_{p,T}^{o}=-RT\ln K_{eq}^{o}=\sum \nu _{i}\mu _{i}^{o}}Где μ o {\displaystyle \mu ^{o}} - стандартные химические потенциалы
Уравнение изотермы показывает, как величина Q связана с изменением свободной энергии реакции:
При
Q
>
K
{\displaystyle Q>K}
для прямой реакции
Δ
G
>
0
{\displaystyle \Delta G>0}
, то есть
∑
ν
j
μ
j
{\displaystyle \sum \nu _{j}\mu _{j}}
для продуктов прямой реакции больше, чем для исходных веществ - это означает, что прямая реакция запрещена (значит, не запрещена обратная);
при
Q
=
K
{\displaystyle Q=K}
для прямой реакции
Δ
G
=
0
{\displaystyle \Delta G=0}
, то есть реакция достигла равновесного состояния;
при
Q
<
K
{\displaystyle Q
Величина по определению имеет смысл только для состояния равновесия, то есть для состояния с v 1 v − 1 = 1 {\displaystyle {\frac {v_{1}}{v_{-1}}}=1} и Δ G r = 0 {\displaystyle \Delta G_{r}=0} . Величина K e q {\displaystyle K_{eq}} ничего не говорит о скоростях реакций, но она описывает состав системы в состоянии равновесия.
Если K >> 1, то в системе преобладают продукты (прямой) реакции Если K << 1, то в системе преобладают исходные вещества (продукты обратной реакции)
Стандартные состояния
Стандартная энергия Гиббса реакции в газовой смеси - энергия Гиббса реакции при стандартных парциальных давлениях всех компонентов, равных 0,1013 МПа (1 атм). Стандартная энергия Гиббса реакции в растворе - энергия Гиббса при стандартном состоянии раствора, за которое принимают гипотетический раствор со свойствами предельно разбавленного раствора , но с концентрацией всех реагентов, равной единице. Для чистого вещества и жидкости стандартная энергия Гиббса совпадает с энергией Гиббса образования этих веществ. Величина стандартной энергии Гиббса реакции может быть использована для приближенной оценки термодинамической возможности протекания реакции в данном направлении, если начальные условия не сильно отличаются от стандартных. Кроме того, сравнивая величины стандартной энергии Гиббса нескольких реакций, можно выбрать наиболее предпочтительные, для которых имеет наибольшую по модулю отрицательную величину.
Кинетическое описание
Для обратимой химической реакции константа равновесия K eq может быть выражена через константы скорости прямых и обратных реакций. Рассмотрим элементарную обратимую химическую реакцию первого порядка
A ⇄ B {\displaystyle \mathrm {A} \rightleftarrows \mathrm {B} }По определению, равновесие задаётся условием v 1 = v − 1 {\displaystyle v_{1}=v_{-1}} , то есть равенством скоростей прямой и обратной реакций.
В соответствии с законом действующих масс v = k ∏ a j n j {\displaystyle v=k{\prod }{a_{j}}^{n_{j}}}
Где k - константа скорости соответствующей реакции, а a j n j {\displaystyle {a_{j}}^{n_{j}}} - равновесные активности реагентов этой реакции, возведённые в степени, равные их стехиометрическим коэффициентам
можно записать условие равновесия в виде
1 = v 1 v − 1 = k 1 ∏ a A n A k − 1 ∏ a B n B {\displaystyle 1={\frac {v_{1}}{v_{-1}}}={\frac {k_{1}{\prod }{a_{A}}^{n_{A}}}{k_{-1}{\prod }{a_{B}}^{n_{B}}}}} 1 = k 1 k − 1 ⋅ ∏ a A n A ∏ a B n B = k 1 k − 1 ⋅ (K e q) − 1 {\displaystyle 1={\frac {k_{1}}{k_{-1}}}\cdot {\frac {\prod {a_{A}}^{n_{A}}}{\prod {a_{B}}^{n_{B}}}}={\frac {k_{1}}{k_{-1}}}\cdot \left(K_{eq}\right)^{-1}}(см. термодинамическое описание константы равновесия), что возможно только если
K e q = k 1 k − 1 {\displaystyle K_{eq}={\frac {k_{1}}{k_{-1}}}}Это важное соотношение даёт одну из «точек соприкосновения» химической кинетики и химической термодинамики .
Множественные равновесия
В случае, когда в системе устанавливается сразу несколько равновесий (то есть одновременного или последовательного протекает нескольких процессов), каждый из них может быть охарактеризован своей константой равновесия, из которых можно выразить общую константу равновесия для всей совокупности процессов. Можно рассмотреть такую ситуацию на примере ступенчатой диссоциации двухосновной кислоты H 2 A. Водный раствор её будет содержать частицы (сольватированные) H + , H 2 A, HA - and A 2- . Процесс диссоциации протекает в две ступени:
H 2 A ⇌ H A − + H + : K 1 = [ H A − ] [ H + ] [ H 2 A ] {\displaystyle H_{2}A\rightleftharpoons HA^{-}+H^{+}:K_{1}={\frac {}{}}} H A − ⇌ A 2 − + H + : K 2 = [ A 2 − ] [ H + ] [ H A − ] {\displaystyle HA^{-}\rightleftharpoons A^{2-}+H^{+}:K_{2}={\frac {}{}}}K 1 и K 2 - константы первой и второй ступеней диссоциации соответственно. Из них можно выразить "полную" константу равновесия, для процесса полной диссоциации :
H 2 A ⇌ A 2 − + 2 H + : K 1 + 2 = [ A 2 − ] [ H + ] 2 [ H 2 A ] = K 1 K 2 {\displaystyle H_{2}A\rightleftharpoons A^{2-}+2H^{+}:K_{1+2}={\frac {^{2}}{}}=K_{1}K_{2}}Другой пример множественного равновесия - анализ системы осадок /растворимое комплексное соединение . Допустим, имеется равновесие
A g I 2 − (a q) ⇌ A g I (s o l i d) + I − (a q) {\displaystyle AgI_{2}^{-}(aq)\rightleftharpoons AgI(solid)+I^{-}(aq)}Реакцию можно представить в виде двух последовательных равновесий - равновесия разложения комплексного иона на составляющие его ионы, которое характеризуется "константой нестойкости" (величина, обратная "константе устойчивости" β):
A g I 2 − (a q) ⇌ A g + (a q) + 2 I − (a q) : K 1 = α A g + α I − 2 α A g I 2 − = β − 1 {\displaystyle AgI_{2}^{-}(aq)\rightleftharpoons Ag^{+}(aq)+2I^{-}(aq):K_{1}={\frac {\alpha _{Ag^{+}}\alpha _{I^{-}}^{2}}{\alpha _{AgI_{2}^{-}}}}=\beta ^{-1}}и равновесия перехода ионов из объёма растворителя в кристаллическую решётку
A g + (a q) + I − (a q) ⇌ A g I (s o l i d) : K 2 = α A g I α A g + α I − {\displaystyle Ag^{+}(aq)+I^{-}(aq)\rightleftharpoons AgI(solid):K_{2}={\frac {\alpha _{AgI}}{\alpha _{Ag^{+}}\alpha _{I^{-}}}}}с учётом того, что для твёрдых веществ активность принимается равной 1 , а в разбавленных растворах активности могут быть заменены на молярные концентрации, получаем
K 2 = α A g I α A g + α I − = 1 [ A g + ] [ I − ] = 1 K s p {\displaystyle K_{2}={\frac {\alpha _{AgI}}{\alpha _{Ag^{+}}\alpha _{I^{-}}}}={\frac {1}{}}={\frac {1}{K_{sp}}}}где K s p {\displaystyle K_{sp}} - произведение растворимости
Тогда суммарное равновесие будет описываться константой
A g I 2 − (a q) ⇌ A g I (s o l i d) + I − (a q) : K = α A g I α I − α A g I 2 − = K 1 ⋅ K 2 = 1 β ⋅ K s p {\displaystyle AgI_{2}^{-}(aq)\rightleftharpoons AgI(solid)+I^{-}(aq):K={\frac {\alpha _{AgI}\alpha _{I^{-}}}{\alpha _{AgI_{2}^{-}}}}=K_{1}\cdot K_{2}={\frac {1}{\beta \cdot K_{sp}}}}И значение этой константы будет условием преобладания в равновесной смеси комплексного соединения или твёрдой соли: как и выше, если K << 1, то в равновесной смеси большая часть ионов связана в комплексное соединение, если K >> 1, то в равновесном состоянии в системе большая часть ионов связана в кристаллической фазе. реакции, протекающей, соответственно, при постоянном давлении или при постоянном объёме. Если Δ H > 0 {\displaystyle \Delta H>0} (тепловой эффект положителен, реакция эндотермическая), то температурный коэффициент константы равновесия d ln K p d T {\displaystyle {\frac {d\ln K_{p}}{dT}}} тоже положителен, то есть с ростом температуры константа равновесия эндотермической реакции увеличивается, равновесие сдвигается вправо (что вполне согласуется с принципом Ле Шателье).
Методы расчета константы равновесия
Расчётные методы определения константы равновесия реакции обычно сводятся к вычислению тем или иным способом стандартного изменения энергии Гиббса в ходе реакции (ΔG 0 ), а затем использованию формулы:
Δ G 0 = − R T ln K 0 {\displaystyle \Delta G^{0}=-RT\ln K^{0}} , где R {\displaystyle R} - универсальная газовая постоянная .При этом следует помнить, что энергия Гиббса - функция состояния системы, то есть она не зависит от пути процесса, от механизма реакции, а определяется лишь начальным и конечным состояниями системы. Следовательно, если непосредственное определение или расчёт ΔG 0 для некоторой реакции по каким-либо причинам затруднены, можно подобрать такие промежуточные реакции, для которых ΔG 0 известно или может быть легко определено, и суммирование которых даст рассматриваемую реакцию (см. Закон Гесса). В частности, в качестве таких промежуточных реакций часто используют реакции образования соединений из элементов.
Энтропийный расчёт изменения энергии Гиббса и константы равновесия реакции
Энтропийный метод расчёта ΔG реакции является одним из самых распространённых и удобных . Он основан на соотношении:
Δ G T = Δ H T − T Δ S T {\displaystyle \Delta G_{T}=\Delta H_{T}-T\Delta S_{T}}или, соответственно, для стандартного изменения энергии Гиббса:
Δ G T 0 = Δ H T 0 − T Δ S T 0 {\displaystyle \Delta G_{T}^{0}=\Delta H_{T}^{0}-T\Delta S_{T}^{0}}Здесь ΔH 0 при постоянных давлении и температуре равно тепловому эффекту реакции, методы расчёта и экспериментального определения которого известны - см., например, уравнение Кирхгофа :
Δ H T 0 = Δ H 298 0 + ∫ 298 T Δ C p d T {\displaystyle \Delta H_{T}^{0}=\Delta H_{298}^{0}+\int _{298}^{T}\Delta C_{p}dT}Необходимо получить изменение энтропии в ходе реакции. Эта задача может быть решена несколькими способами, например:
- По термическим данным - с опорой на тепловую теорему Нернста и с использованием сведений о температурной зависимости теплоёмкости участников реакции. Например, для веществ, при нормальных условиях находящихся в твёрдом состоянии:
Итак, если известны Δ H 298 0 {\displaystyle \Delta H_{298}^{0}} , Δ S 298 0 {\displaystyle \Delta S_{298}^{0}} и температурные зависимости теплоёмкости, Δ G T 0 {\displaystyle \Delta G_{T}^{0}} может быть рассчитано по формуле:
Δ G T 0 = Δ H 298 0 − T Δ S 298 0 + ∫ 298 T Δ C p d T − T ∫ 298 T Δ C p d T T {\displaystyle \Delta G_{T}^{0}=\Delta H_{298}^{0}-T\Delta S_{298}^{0}+\int _{298}^{T}\Delta C_{p}dT-T\int _{298}^{T}\Delta C_{p}{\frac {dT}{T}}}Несколько упрощённый вариант этой формулы получают, считая сумму теплоёмкостей веществ не зависящей от температуры и равной сумме теплоёмкостей при 298 K:
Δ G T 0 = Δ H 298 0 − T Δ S 298 0 + Δ C p 298 (T − 298) − T ln T 298 {\displaystyle \Delta G_{T}^{0}=\Delta H_{298}^{0}-T\Delta S_{298}^{0}+\Delta C_{p~298}(T-298)-T\ln {\frac {T}{298}}}И еще более упрощённый расчёт проводят, приравнивая сумму теплоёмкостей к нулю:
Δ G T 0 = Δ H 298 0 − T Δ S 298 0 {\displaystyle \Delta G_{T}^{0}=\Delta H_{298}^{0}-T\Delta S_{298}^{0}}Переход от Δ G T 0 {\displaystyle \Delta G_{T}^{0}} к константе равновесия осуществляется по приведённой выше формуле.
